Important Distinctions Among USP 1115, USP 1116, and USP 1211
What is medical device and product environmental monitoring?
The methods and instruments used to observe an environment, describe its quality, and make sure it satisfies predetermined acceptance standards are known as environmental monitoring. Environmental monitoring for medical devices and products includes acceptability requirements for the environment at every stage of the product’s lifecycle, from raw materials to expiration or end-use. One crucial parameter for quality control is environmental monitoring. Verifying that your product or medical device is free of microorganisms and that the environment during the various stages of production, packaging, shipping, and storage has minimal exposure to microorganisms is especially important. Environmental monitoring to maintain production conditions for non-sterile and aseptic operations is covered in USP 1115, USP 1116, and USP 1211 SAL (sterility assurance levels).
What does USP 1115 cover?
The bioburden control of non-sterile pharmacological ingredients and products is covered by USP 1115. With regard to bioburden requirements, USP 1115 will assist you if your product is not sterile.
What does USP 1116 cover?
The microbiological monitoring and control of aseptic processing environments is covered by USP 1116. USP 1116 will help you with microbiological control and regulations if your product needs to be manufactured aseptically.
What does USP 1211 cover?
Sterile assurance is covered under USP 1211. For both sterile and non-sterile items that are produced aseptically or go through terminal sterilization procedures, USP 1211 is crucial.
What distinguishes USP 1115, USP 1116, and USP 1211 from one another?
Regulations pertaining to bioburden and sterility assurance are covered by USPs 1115, 1116, and 1211. Nonetheless, the components discussed in USP 1115, USP 1116, and USP 1211 differ significantly. Non-sterile monitoring and production circumstances covered by USP 1115 are exempt from the same quality standards and limitations as the aseptic manufacturing conditions covered by USP 1116. Terminal sterilization, the third kind of sterilizing procedure, is covered by USP 1211. By a final sterilization process that ensures a measurable safety level, the majority of terminally sterilized products meet the same sterility requirements as aseptically treated products without going through aseptic processing. Aseptic processing is far more expensive than terminal sterilization. Therefore, rather than using aseptic processing, the majority of items will use non-sterile manufacturing followed by terminal sterilization to meet FDA sterility criteria.
Summary of Nonsterile Product Processing (USP 1115)
Non-sterile processes: what are they?
Non-sterile processes are techniques or operations carried out in a setting where bioburden is managed to acceptable levels according to the characteristics of the product, the administration route, and the intended patient population. In contrast to sterile processes, which eradicate the bioburden, non-sterile processes do not. The following list of non-sterile products is arranged from high to low according to the possible danger of microbial contamination. Medical equipment intended for usage in the same bodily parts (mouth, rectum, skin, vagina, and nasopharynx) are included in the same list.
A list of typical pharmaceutical items that are not sterile:
- Dry powder and metered-dose inhalants
- Sprays for the nose
- The field of optics
- Suppositories for vagina
- Topicals
- Reproductive suppositories
- Aqueous solutions taken orally
- Capsules filled with liquid
- Powder-filled capsules and oral tablets
Why does your medical device or product need environmental monitoring for non-sterile processes?
In contrast to sterile products, nonsterile products have their microbiological content regulated to the amount required for patient safety within the product’s use restrictions. Overly strict sterility or aseptic processing regulations increase expenses and complexity without improving safety. Manufacturers and patients both save money when superfluous sterility or aseptic processing restrictions are removed. Depending on how your medical product is going to be used, non-sterile processing techniques might be beneficial. The processes needed to create nonsterile products and manage product microorganisms differ greatly from those needed for sterile products. Sterile products are given topically or injected into delicate tissues that have no barriers to infection, little or no microbial flora, and a high risk of infection. Nonsterile products, on the other hand, are applied to areas of the human body that have strong natural microbial flora densities together with immunological and physical defenses against infection. Non-sterile products must nevertheless undergo industrial processing to avoid excessive contamination, despite the tighter microbiological restrictions for sterile products. One of the biggest concerns for non-sterile goods is microbial growth in excipients, components, and medicinal ingredients, which is closely monitored. Below is a collection of typical non-sterile product examples.
How are non-sterile product manufacturing settings monitored?
One qualitative method for reducing the risk of contamination in non-sterile products is the monitoring of industrial surroundings for microorganisms. A robust environmental monitoring program saves time and money by confirming the efficacy of microbiological controls and identifying unanticipated contamination issues early. Although they can be applied in aseptic facilities, microbial procedures and methods are not meant for nonsterile settings. The degree of human activity and the gowning requirements of the industrial facility frequently determine the amounts of temporary contamination. Since people are the primary source of microbiological contamination in clean surroundings, the aforementioned is accurate. In nonsterile items, manufacturers anticipate regulated bioburden levels that won’t endanger the consumer. To maintain the efficacy of the facility’s microbial environmental controls, producers must determine acceptable quantities of microorganisms in each product and conduct routine production plant hygiene assessments. Microbial sampling, staff evaluations for proper gowning and SOP execution, raw material assessments, and cleaning protocol efficacy evaluations are all examples of hygiene assessments.
Microbial monitoring is not necessary once a product has been put into a packaging container. For non-sterile manufacturing, products with microbiocidal properties or resistance to microbial colonization will require little to no microbiological monitoring. Furthermore, settings used in the production of tablets, powders, and liquid-filled capsules don’t require regular or any monitoring. The possible risk connected to the dosage form is reflected in the frequency of microbiological monitoring. Usually created in ISO level 8 classed rooms, manufacturing spaces for higher-risk dosage forms, like inhalant medicines, need more frequent monitoring.

Summary of Aseptic Product Processing (USP 1116)
Describe aseptic procedures.
Methods or procedures used in a sterile setting (such an isolator) are known as aseptic processes. Specialized tools that stop microbiological material from technicians, raw materials, or machinery from contaminating medical devices or goods help preserve the aseptic sterile environment.
Sterile and aseptic are not interchangeable terms. The methods by which microbial contamination is avoided are different for sterile and aseptic items, but both will stop it after use. A complete lack of living microorganisms with the capacity to proliferate is referred to as sterile. Therefore, after being put in their final packing, sterile products are frequently thermally or chemically sterilized. Any bacteria found within the products (obtained during manufacturing and packaging) are eliminated by chemical or heat sterilization. Terminal sterilization is the process of sterilizing a product using heat or chemicals after it has been packaged.
However, by keeping microorganisms out, an aseptic procedure avoids contamination. Sterile and aseptic are used interchangeably, despite the fact that they have different definitions. In fact, aseptic processing, as opposed to terminal sterilization, is used in the manufacturing of many sterile items.
What Kinds of Medical Products Are Produced in Aseptic Settings?
- Sterile pharmaceutical items
- Sterilized drug compounds in bulk
- Intermediates that are sterile
- Excipients
- Medical equipment
- The biologics
Only ISO-classified clean environments, restricted-access barrier systems (RABS), and isolators utilized for aseptic processing should be subject to the USP guidelines cited for microbiological examination. The levels of contamination control necessary for sterile items manufactured aseptically do not need to be met in other clean environments. Refer to USP 1115 for goods that need non-sterile processing, such as nasal sprays, topicals, or oral tablets.
Why Does Your Medical Device or Product Need Environmental Monitoring for Aseptic Processes?
The removal of microorganisms from the manufacturing process is necessary for aseptic processing. During processing, open containers and product components must be kept free of microorganisms. Therefore, the risk of intolerable microbiological contamination is influenced by both the bioburden of the product and the bioburden of the manufacturing environment. Aseptic processing surroundings must be kept free or to a minimum of microorganisms to reduce the danger of patient infection after product usage, since aseptic processing goods will not be terminally sterilized. Cleanroom clothing is not required nor allowed for operators performing sophisticated aseptic procedures.
What Should Be Considered When Monitoring the Environment for Aseptic Procedures?
Microbiological contamination is unavoidable in settings where people are operating. If there are human operators present, even the most meticulous operation and careful clean-room environment design cannot completely prevent the shedding of microorganisms. It is therefore impractical to aim for zero contamination at aseptic sites throughout each aseptic procedure.
There are no foolproof ways to make sure that an aseptic processing environment and the surfaces that come into contact with the product are always sterile. In a technical sense, monitoring findings cannot confirm or deny sterility. To ensure that the facility runs in a verified state of control, producers should regularly check the findings of environmental monitoring.
Manufacturers are unable to guarantee a sterility level by monitoring, statistics, or aseptic processing simulations due to monitoring restrictions. The best way to ensure sterility is to concentrate on human-borne contamination and design the facility to reduce the danger of this contamination.
Reducing or eliminating human interventions through appropriate equipment design and by providing enough air exchanges per hour for the facility’s staff population significantly reduces the danger of microbial contamination. Other contamination risk mitigation strategies that can break the chain of infection include efficient people and material mobility, appropriate temperature and humidity control, and routine sanitization. In different aseptic conditions, the recommended contamination recovery rates are listed in Table 3 of USP 1116 (reproduced as Table 1 below).
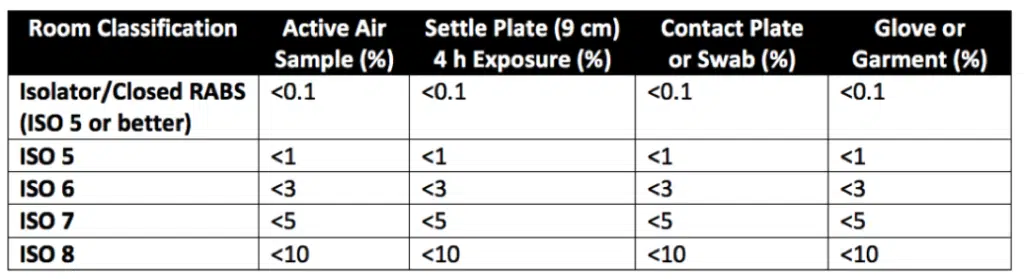
Table 1. ISO Cleanroom Classification’s Recommended Contamination Recovery Rates for Aseptic Environments
Summary of USP 1211 Sterility Assurance
Sterility assurance: What Is It?
Aseptic processing, post-aseptic fill terminal sterilization, and terminal sterilization are all included in sterility assurance, which includes environmental monitoring.
Terminal Sterilization: What Is It?
Unlike aseptically made products, terminally sterilized products undergo a final sterilization step that ensures a measurable safety level. Therefore, terminally sterilized products are the least risky sterile medical products in terms of microbiological risk. Heat-steam sterilization is the most common method used for terminal sterilization. However, terminal sterilization can also be accomplished by irradiation (e.g., gamma or e-beam technology) and chemical sterilization. The probability of non-sterility (PNS) is the measure of sterility assurance for products that have undergone terminal sterilization. A PNSU of ≤ 1,000,000, or the likelihood of producing no more than one nonsterile unit per million, must be attained using terminal sterilization procedures.
What is Sterilization of Post-Aseptic Processing Terminals?
Aseptically manufactured products can control the pre-sterilization bioburden, which can make the subsequent terminal sterilization processing less harsh for the product or medical device. This can be achieved by exposing the product to lower temperatures, shorter cycle times, or less chemicals for the final sterilization process. Post-aseptic processing terminal sterilization is the term used to describe terminal sterilization of aseptically manufactured products, where the goal of the terminal sterilization cycle is to kill low bioburden microorganisms rather than pass biological indicators.
How Do You Carry Out Sterility Assurance Environment Monitoring?
Environmental monitoring for sterility assurance qualitatively assesses the effectiveness of a facility’s design and operational controls to provide sterile products. As detailed in Figure 1 below, many external factors influence product sterility. While environmental monitoring is important, it cannot substitute for good facility equipment, process design, and practices. Indeed, monitory only provides a snapshot of the actual environmental conditions occurring moment-to-moment in a manufacturing facility. Additionally, excessive environmental sampling can impair product safety and performance in critical anti-microbial areas. There are inherent limitations with all viable and non-viable monitoring forms in terms of sample size, sample location, and recovery capability. Sterility testing and aseptic process simulations also have their limits. However, viable monitoring, non-viable monitoring, aseptic process simulations, and sterility testing are all excellent methods to perform environmental monitoring for sterility assurance of manufactured medical devices and products. These techniques ensure that established performance criteria, according to the ISO classification of the room, are met.
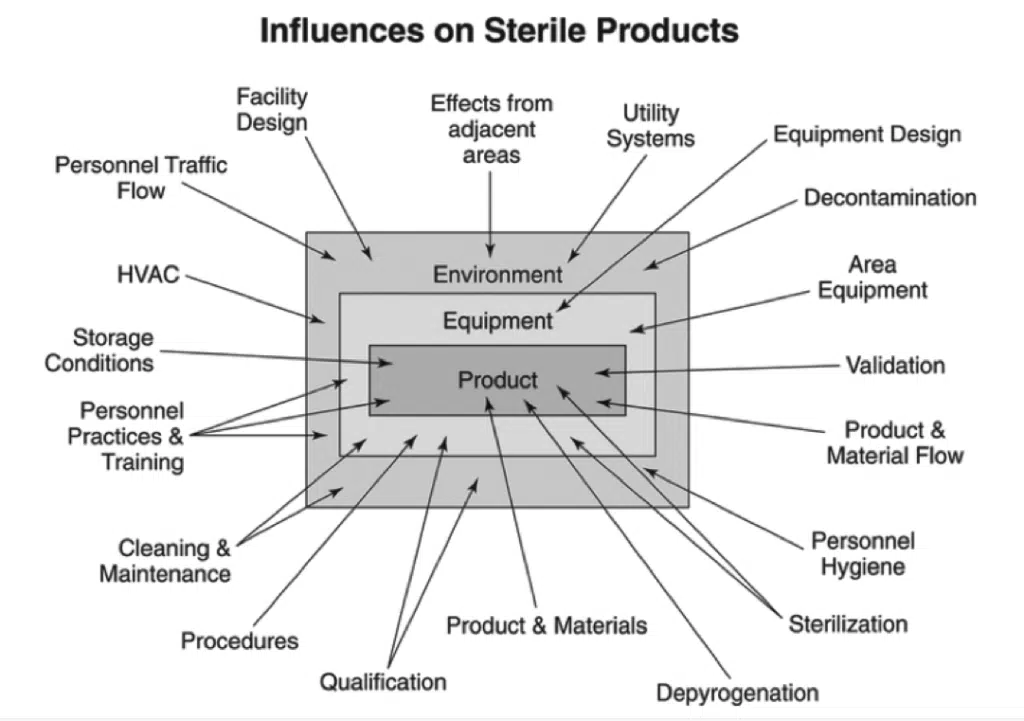
Figure 1: External factors affecting sterile goods
Practical Surveillance
Microbial sample methods are used in viable monitoring to identify and quantify the amount of culturable microorganisms present in the air, on surfaces, and on people.
Practical monitoring sampling techniques are:
- Air sampling that is active
- sample of the air passively
- Use of fluorescence technology for practical particle counting
- An examination of surfaces, gloves, and gowns using contact plates
- Surface swabbing
- Monitoring of personnel
Non-Viable Surveillance
Non-viable monitoring uses calibrated particle counters to measure the quantity and size of particles (dead or alive) in the air. The cleanroom is first categorized using ISO 14644-1 non-viable monitoring, and normal manufacturing conditions are periodically evaluated.
Simulations of Aseptic Processes
Process simulations use a sterile growth medium to assess how well an aseptic activity performs. The product can be added to or immediately replaced with the sterile growing medium. Process simulators accurately depict the circumstances and operations of production processing, whether they are new, updated, or existing. Before introducing a new or changed process component (new product, facility, equipment, personnel, etc.), aseptic process simulations are frequently carried out to determine whether the addition satisfies aseptic performance requirements.
Testing for Sterility
To ascertain whether the material’s requirements are fulfilled, sterility testing is carried out. Finished items must undergo lot-by-lot sterility testing unless parametric release testing is authorized. One sterility assurance release program that shows control over the sterilizing process for reliable lot-by-lot sterility outcomes is the parametric release. The most popular method of releasing sterile products at the moment is parametric release.
In brief
In general, contamination control for aseptically created products (USP 1116) is different from contamination control for nonsterile product manufacturing (USP 1115). Additional requirements are also met by items that go through sterility assurance for terminal sterilization following nonsterile or aseptic processing (USP 1211). Products that are sterile and aseptically produced are applied or injected into delicate tissues that have little to no microbial flora, no barriers to infection, and a significant risk of infection. Nonsterile products, on the other hand, are applied to areas of the human body that have strong natural microbial flora densities together with immunological and physical defenses against infection. Oral tablets, nasal sprays, topicals, and rectal suppositories are a few examples of non-sterile products. The microbiological content of nonsterile items is regulated to the amount required for patient safety within the product use parameters, in contrast to sterile products, which have a fixed microbial limit. Manufacturers and patients both save money when superfluous sterility or aseptic processing restrictions are removed. Overall, make sure you pick a contract testing company that can assist you with the proper sterility testing, sterilization validations, and environmental monitoring for your particular medical device or product needs, regardless of whether you require sterile or non-sterile processes.





