What Is the Price of Developing Medical Devices?

What are medical devices?
Numerous nonpharmaceutical products used for illness treatment, monitoring, or prevention are categorized as medical devices. For instance, the concept of a medical device includes mechanical devices, combination devices, and medical instruments (electronics). Certain combination devices contain biological, pharmacological, or chemical ingredients. Medical devices also include surgical instruments, implanted devices, and non-implantable gadgets that patients use at home. Given the abundance of medical devices available on the market, their significance for hospitals and the healthcare sector as a whole cannot be overstated. Medical device definition, benefits of medical device development companies, medical device development procedures, medical device application fees, and medical device costs for FDA clearance are all covered in this article.
Does the US Food & Drug Administration (FDA) classify my product as a medical device?
The FDA regulates all medical devices. You can find examples of manufacturers who are required to list their devices with the FDA HERE. The primary basis for your product’s eligibility as a medical device is its intended purpose and usage indications. A medical device is defined by the Food, Drug, and Cosmetic Act as:
“An instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory that is: 1) recognized in the United States Pharmacopoeia or the official National Formulary, or any supplement to them, 2) intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or 3) intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals, and which is not dependent upon being metabolized in order to achieve its primary intended purposes. Software features that are prohibited by section 520(o) are not included in the definition of “device.”
Which medical devices are classified by the FDA?
Depending on the possible risk to human health during intended use, the FDA classifies devices into one of three regulatory classifications. Class I devices pose little or no risk of injury to people. Compared to Class I devices, Class II devices are riskier. Class III devices include implantable devices, life-sustaining or life-supporting equipment, and devices that carry a significant risk of disease or harm when in use. All that is needed for Class I and II devices is a 510(k) submission. For FDA marketing clearance, Class III devices must submit a complete premarket approval (PMA) application. Certain Class I and Class II devices are exempt from 510(k) filing requirements. The exemptions granted to all exempt devices are subject to restrictions. 21 CFR Parts 862-892 provide details on the restrictions on device exemptions. Furthermore, Class III devices that are essentially identical or that were sold before to the FDA’s 1976 modifications (pre-amendment devices) only need to submit a 510(k) rather than a full PMA. Note that a complete PMA will be needed for marketing approval of pre-amendment devices with “significant changes.” Both the device’s intended application and its indications for use are taken into account when submitting a 510(k) vs a PMA for Class III substantially identical devices. Table 1 below (reproduced from a presentation by Kimberly Piermatteo) and our article on FDA medical device classifications provide more information on Class I, Class II, and Class III devices as well as the approximate percentage of medical devices developed by companies in each Class (either through their own medical device development processes or through outsourcing to a medical device development company).

Table 1: FDA Classifications of Medical Devices by Device Risk and Types of Required Submissions
What is the manufacturing cost of medical devices?
The expenses associated with bringing a novel medical equipment to market vary depending on the type of device being sold (Class I, Class II, or Class III) and its intended purpose. The amount of money required to finance a novel medical equipment depends on a number of factors. The kinds of medical device development procedures needed for the new device are among these variables. For instance, if a new device needs clinical studies, biocompatibility and quality control testing, and has a high level of medical device technology complexity (or novelty), a medical device start-up might need additional funding and get in touch with a medical device development company, contract testing company, and clinical studies coordinators.

A study of 750 of the 1,000 medical device businesses in the market was used to gather and publish data in 2010. The average overall cost to get a Class II 510(k) product from concept to FDA clearance was $31 million, according to data from the 200 companies who replied. About $24 million of this $31 million was allocated to duties that were related to or dependent on the FDA. Of the $31 million, an additional $2 to $5 million was used for initial engineering and development expenses. Typically, 12% of the entire cost of bringing a 510(k) medical device to market goes toward engineering and development. The $2–$5 million in development costs approaches 30% of the overall cost of the medical device project when clinical studies are not required.
On the other hand, it typically cost $94 million to bring a Class III medical device from idea to premarket approval (PMA). About $75 million of this $94 million was allocated to activities that were tied to or dependent on the FDA. A chart of the money raised for different medical equipment may be found in Table 2 below. As one might anticipate, Class III medical devices—those that required clinical trials—required a lot more funding. Additional equity can be required for businesses that have not received regulatory clearance in order to address clinical results, finish production transfers, and complete scale-up.
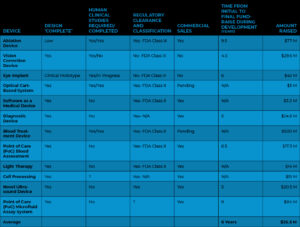
Table 2: Funds Raised for Different Medical Equipment
On average, the 2010 study required 20 months for proof of concept and beginning development, 12 months for clinical development, and 40 months for 510(k) approval. A 510(k) medical device took roughly six years to go from beginning development to clearance. At 12 years, PMA medical device timelines were around twice as long as 510 (k) products.
What is the cost of FDA fees for new applications for medical devices?
You may find the FDA’s current medical device application fees here.
Table 3 below is a copy of the FDA fee schedule for 2022, as of October 2021. A startup is likely to be eligible for a small company charge. The Center for Devices and Radiological Health (CDRH) must certify a company as a small business before it can submit small business payments. Typically, small business rates range from half to a quarter of the FDA’s regular expenses.
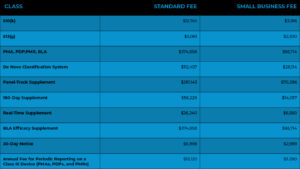
Table 3: FDA 2022 Schedule of Application Fees for Medical Devices
In Brief
A wide variety of nonpharmaceutical products used for illness treatment, monitoring, or prevention are referred to as medical devices. Certain combination devices contain biological, pharmacological, or chemical ingredients. Medical devices also include surgical instruments, implanted devices, and non-implantable gadgets that patients use at home. Depending on the possible risk to human health during intended use, the FDA classifies devices into one of three regulatory classifications. Class I devices pose little or no risk of injury to people. Compared to Class I devices, Class II devices are riskier. Class III devices include implantable devices, life-sustaining or life-supporting equipment, and devices that carry a significant risk of disease or harm when in use. All that is needed for Class I and II devices is a 510(k) submission. For FDA marketing clearance, Class III devices must submit a complete premarket approval (PMA) application. In 2010, the average total cost from concept to FDA clearance for a Class II 510(k) product was $31 million. On the other hand, it typically cost $94 million to bring a Class III medical device from idea to premarket approval (PMA). After accounting for inflation, these figures rise by about 20% between 2010 and 2020, reaching $37.2 million for Class II 510(k) devices and $112.8 million for Class III PMA devices. A 510(k) medical device took roughly six years to go from beginning development to clearance. At 12 years, PMA medical device timelines were around twice as long as 510 (k) products. All things considered, whether you are just starting out with medical devices or are a few years into the process, make sure you pick a contract testing company that can help you with the right biocompatibility testing for your product, implant, or medical device.





